How to use Vsfold5 on the web
General Information:
For the simplest calculation where defaults are all that is required:
1. Write a name for the sequence. Because this makes a file, you should not use any special characters, only Roman characters, ‘-‘, ‘_’ and ‘ ‘ (space) will be processed.
2. Enter a valid email address. When your calculation is finished, you will be emailed a link to the results so you can download any data you want or need from the calculation.
3. Enter the sequence you want to calculate. Currently, there is an upper limit of 450 nt because of time out issues on the server.
4. Go to the bottom of the page and press [Submit Query]
An example input is shown in Figure 1.

Figure 1: Relevant parts of the most basic submission for a query.
Basic options
Most sequences require some thinking on the part of the user before clicking the [Submit Query] button. RNA serves different purposes. The default settings are intended to be applied to small tightly folded structures like tRNA and some rather moderately small and compact pseudoknots. Functional RNA tends to have scaffolding that makes it rather stiff. Such structures require a larger Kuhn length (typically 10 nt) where the Kuhn length is a measure of the stiffness of the RNA. Therefore, for many important RNA structures, one needs to consider what their purpose is and where in the life cycle they appear.
Three examples of fitting pseudoknots are offered here for reference: tRNA(phe), the Upsteam pseudoknot (UPSK), and the Ribosomal S15 leader sequence.
Fitting tRNA(phe):
This is a structure that satisfies all the default input values. It also just happens to have a D and T loop region that form a nice solid stem of standard Watson-Crick base pairs. All the default settings are used, and one only has to click [Submit Query] to obtain the desired result.
|
|
|
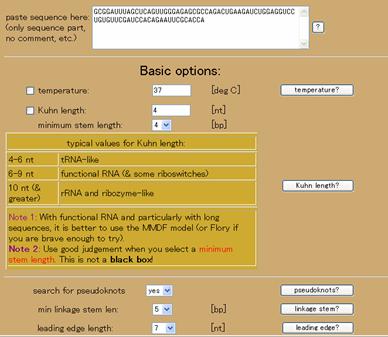
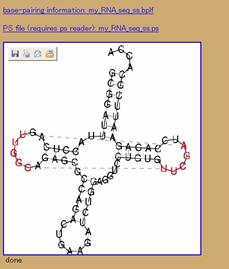
Figure 2: An example of using the default settings to catch tRNA(phe). (a) Using default settings. (b) the resulting structure.
The Upsteam pseudoknot (UPSK)
The sequence is available at http://www.sanger.ac.uk/cgi-bin/Rfam/getacc?RF00390. It is a very small pseudoknot and therefore, the default parameters are not able to catch it. The parameters to be changed are indicated in green. To find very compact and tightly wound pseudoknots like this, the minimum stem length should be reduced to 3 bp (note in Figure 3b that one of the stems is 3 bp long!). Likewise, currently, the default search for pseudoknots generally ignores stems shorter than 5 bps because the bonds are typically too feeble to take seriously with current parameterization. However, when you know that there are such tightly compact structures, or if you want to know if any such structures as these exist, then you must change the minimum linkage stem length accordingly. Following the settings of the green circles in Figure 3a, the result is shown in Figure 3b.
|
|
|
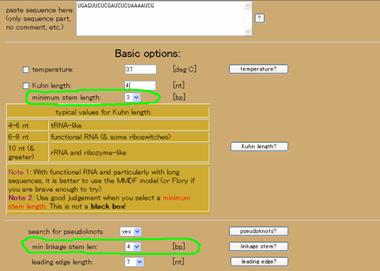
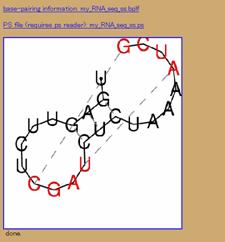
Figure 3. Example of fitting the Upsteam pseudoknot (UPSK). (a) Changes to the default settings. (b) The results.
Ribosomal S15 leader:
This sequence can be found at http://www.sanger.ac.uk/cgi-bin/Rfam/getacc?RF00114. This structure is rather stiff and seems to better fit with a Kuhn length of 8 nt or so. The Kuhn length is a measure of the stiffness. The remaining settings are sufficient at their default settings. As in Figure 4a, change the Kuhn length by clicking the left hand box (toggle) so that the label “Kuhn length” turns red. Then change the value in the box next to it as shown in Figure 4a. The result of the calculation is shown in Figure 4b. Note: the parts of the structure predicted in this example agree with those indicated for this structure; however, there is additional secondary structure and a pseudoknot that are predicted in addition to the consensus structure.
|
|
|
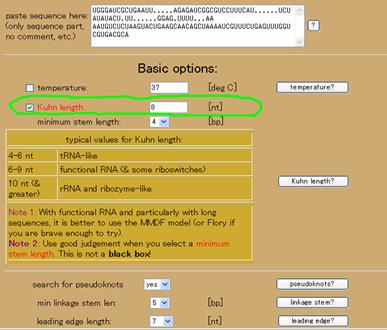
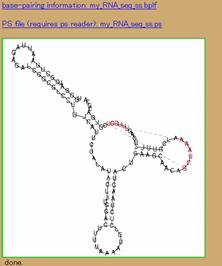
Figure 4. Example of fitting the Ribosomal S15 leader. (a) Changes to the default settings. (b) The results.
Final comments:
Note that the sequence from the Rfam database was cut and pasted into the box. This raises one other point. When inserting sequences into the box, do not include any file information that contains text. All ASCII characters except AaCcGgTtUu will be filtered out automatically.
Note also that not everything in the Rfam data base can be fit using this model. Some reasons are because there are proteins involved in the processes, there are vastly different Kuhn lengths in the structure, limitations of the current vsfold5 version (which will gradually be improved with more testing, experiment, and development), etc. Also, your mileage (kilometers) may vary. The standard disclaimers apply.